The past month has been an amazing time for AI news. I post this amid a flurry of ChatGPT screenshots and at the leading edge of a ton of blog posts on CASP15 results in the coming weeks. But we should be careful not to miss out on a pre-publication that I believe could be one of the most disruptive technologies in the structural biology field in the coming decade.
The Baker Lab—the originator of the Rosetta algorithm for ab initio protein structure prediction and a slew of other protein structure related tools like Rosetta@Home and FoldIt—released a prepublication at the start of this month. There, they introduce RFDiffusion, a novel application of Diffusion algorithms to the protein design problem.
Using RFDiffusion, the authors (Joseph Watson, David Juergens, Nate Bennett, Brian Trippe, and Jason Yim et al.) achieve multiple orders of magnitude advances in de novo protein design. With these advances, I believe we are on the cusp of a new era of protein design where applying proteins to complex pharmaceutical and industrial problems becomes the go-to solution.
The Baker Lab, founded by Dr. David Baker at the University of Washington, has been a leader in the structural biology space for over a decade. The lab has focused on developing novel methods and technologies to answer fundamental questions in the structural biology field. Specifically, the Baker Lab has focused on three main areas: protein structure prediction, protein-protein docking, and protein-drug design.
In the field of protein structure prediction, the Baker Lab has developed a suite of computational tools, such as Rosetta and FoldIt, to predict the structure of proteins from their amino acid sequences. This work has enabled researchers to better understand the functions of proteins and has led to the development of new treatments for a range of diseases.

Protein Structure Prediction
Protein structure prediction was, for a long time, a holy grail of structural biology. Solving how a specific sequence of amino acids would fold into a 3D structure is immensely difficult due to the number of degrees of freedom involved. Physically based folding solutions using molecular dynamics were able to approximate this process to some extent, but at the cost of enormous amounts of compute.
Running a full physical simulation of every atom requires vast amounts of processing time, necessitating solutions like the massively distributed Folding@Home project. This distributed supercomputer achieved a total compute of over 2 exaflops in 2020.
Since 2016, however, significant steps were being made in this space with the application of machine learning to the problem. The CASP competition, which had been running biannually for 13 iterations, finally saw improvement after a decade of relative stagnation. In 2018, Google's DeepMind competed with their initial submission of AlphaFold. This deep learning based system achieved a ~20% improvement over the next competitor.
In 2020, AlphaFold2 swept the board with a 265% score improvement over the Baker Lab's submission. Following this, Science's Robert F. Service called protein structure prediction "Solved by AI".
AlphaFold is a protein folding algorithm developed by DeepMind, a subsidiary of Google. It is designed to predict the 3D structure of a protein based on its amino acid sequence. AlphaFold uses deep learning and other artificial intelligence techniques to accurately predict protein structures, which can help scientists better understand how proteins interact with each other and in turn, open up new avenues of research into the prevention and treatment of diseases. AlphaFold has already achieved remarkable success in the biennial Critical Assessment of Structure Prediction (CASP) competition, outperforming all other competitors and taking the top prize in 2020.
Nevertheless, the frontiers are always shifting. With one problem solved, a multitude of new challenges rear their head, previously not even considered possible to tackle. The Baker Lab is back with a vengeance, applying their own in-house tech to revolutionize protein design in several novel ways.

Diffusion Models
Diffusion models are a relatively new type of model that allow for the generation of complex outputs. Proposed in 2015 as a technique for generating images, these models took a back seat to the emerging GAN architecture. The latter employs two models that are alternately trained in an adversarial manner to generate images and discriminate images generated by the network from those in the training set. This approach was able to create quite realistic looking imagery (consider StyleGAN and thispersondoesnotexist.com), but training the GANs is challenging. Having tried to train these myself back in 2016, balancing the power of the generator and discriminator is important and the long training runs can be unstable. Nevertheless, GANs were able to produce convincing results and were even able to be conditioned into generating different classes and properties when specified.
Diffusion models operate quite differently. Instead of attempting to generate an image on the first try, Diffusion models iteratively improve on a noisy image, denoising the input to some extent with each step. The denoising process can be guided by another neural network, like CLIP, in order to produce results in line with a textual description. The training process is more stable and conditioning is far more fine grained, allowing for a wider range of images that can be generated.
OpenAI CLIP (Contrastive Language-Image Pre-training) is a pre-training technique developed by OpenAI, which uses a contrastive learning approach to learn a joint representation of text and images. The model takes in pairs of text and images, and learns to map them to a common joint representation. This joint representation is then used for downstream tasks such as image captioning, question answering, and more. OpenAI CLIP has shown to outperform existing pre-training models on several tasks.
Diffusion models reached public consciousness in April of 2022, with the release of DALL-E 2 by OpenAI. While the initial release of DALL-E used a VQGAN architecture, the second iteration was equipped with a diffusion based generator. Beta access was granted to select individuals in July, but alternative implementations of the technology began sprouting up even before that. Midjourney became accessible on July 12th and Stable Diffusion, a fully open source implementation, was released in August.
The release of these models resulted in an outpouring of creativity, with over 5 million images recorded by Stable Diffusion search engine Lexica.art as of September.
The general principle of iteratively updating some input to get better results wasn't exactly new in the machine learning structural biology space. AlphaFold implements this method as well in order to make fine grained updates to the output structure after first finding a good approximation using the multiple sequence alignment. It is even hypothesized that this recycling function is a good approximation of the energy function actually used for folding. This process was cited as an inspiration by the authors for the RFDiffusion paper. Work on integrating diffusion with protein structure models for scaffolding has been going on at the Baker Lab at least since the start of this year, and improving on it would lead to the amazing innovation that is the topic of this blog post.

RFDiffusion
On December 1st 2022, Joseph Watson from the Baker Lab used DALL-E as a hook to introduce their newest pre-publication: "Broadly applicable and accurate protein design by integrating structure prediction networks and diffusion generative models". Using diffusion models with protein generation models allowed them to design proteins quickly and accurately. They named their technology RosettaFoldDiffusion (RFDiffusion).
Using the RossettaFold protein folding model with a Diffusion approach allows them to specify some constraints—like a target protein to bind to, or a symmetry type—and generate a protein structure that satisfies those constraints.
On December 10th 2022, David Juergens posted an update on Twitter. Reportedly their experimental results had come in, with 384 different designs straight out of the algorithm being tested for binding affinity. Conservatively, 18% of these 384 proteins showed binding responses of <10uM, a two order of magnitude increase compared to previous Rosetta-based processes. All working proteins were shown to compete with a positive control, meaning that the designs likely targeted the intended region for binding.
Implementation
In the RFDiffusion paper, the authors make use of an SE(3) equivariant diffusion model, referred to as DDPMs, to generate the backbone. Recent work has adapted these DDPMs for protein monomer design, with limited success. To address this, RosettaFold was used as a good candidate to improve on this, as it had already shown that it could design parts of protein backbones by doing inpainting of protein structures, which was then adapted to RFjoint Inpainting to design protein scaffolds in a single step.
RosettaFold was finetuned as a denoiser. To train the model, a training set is generated by simulating the noising process for up to 200 steps on structures from the Protein Data Bank, with both the backbone and the residue orientation being perturbed. The Ca positions are translated randomly and residues are randomly rotated in a stepwise manner, and the model is trained to minimize mean squared error loss between predictions and the true structure.
During inference, a random set of residues are generated and fed into RFDiffusion to denoise. The input for the next step is calculated by moving into the direction of the denoised step and adding some noise, to make sure that the denoising process mimics the initial noising process used for training. This is done iteratively until a protein-like structure is formed. When the backbone is designed, ProteinMPNN is used to predict a sequence that will translate into that backbone. The generated structure is then checked against the output of AlphaFold2 for the designed sequence. If the generated structure is close to the AF2 result, it is deemed a successful design in silico.
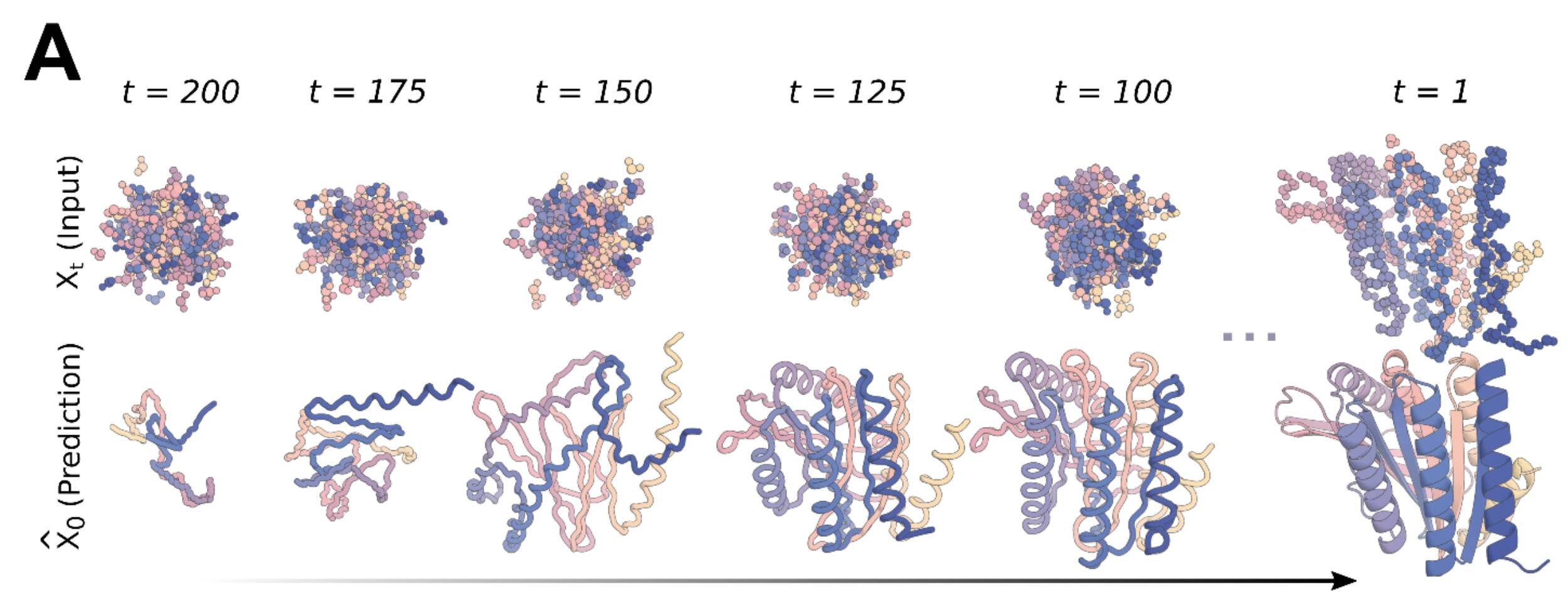
Capabilities
RFDiffusion has turned out to be a powerful tool capable of designing protein structures with a variety of applications:
Unconditional monomer generation: It can generate monomers with little similarity to existing known protein structures. Generating a 100 residue structure takes approximately 2.5 minutes on an A4000 GPU (with time requirements likely scaling quadratically with sequence length). In vitro characterization of 11 TIM barrel designs indicated that nine designs aligned with expected protein parameters.
Higher order oligomers: It can design oligomers that serve as vaccine platforms, delivery vehicles and catalytic agents. The expected oligomerization pattern was found in ~16% of generated structures in vitro.
Motif scaffolding: It can design scaffolds for protein structural motifs used for binding and catalytic functions, with the constraint that the motif must retain its exact geometry for optimal performance. Out of 25 benchmark problems, RFDiffusion solves 23, 11 more than competitor "Hallucination". No tuning was required.
De novo protein binders: Characterized as a grand challenge in protein design, RFDiffusion achieved an 18% success rate in designing protein binders. Experimental success rate compared to Rosetta-based methods ranges from 5x to 214x improvement depending on the target protein. RFDiffusion was finetuned on protein complexes in order to achieve this.
Implications
What is possible with this kind of model combined with its reported effectiveness is nearly endless. If these results hold up in practice, this could easily be the best application of diffusion models for mankind. It could share the crown for biggest advance in structural biology of this decade together with AlphaFold.
Currently RFDiffusion is a potent method for the creation of protein-binders. Applications for this specifically are amazing in themselves, as it will allow for the rapid design and iteration of a myriad of drugs that target proteins specifically. Rapidly designing antibodies comes to mind immediately, provided that the technique proves robust for this notoriously hard to model protein type. Even without antibodies in play, developing inhibitors or activators for any number of proteins would be exponentially sped up by this innovation.
Next steps are already being taken to incorporate nucleic acids into the model, allowing for DNA or RNA binding proteins to be designed rapidly. These types of drugs are used as anti-cancer agents and could yield amazing results in that field.
This is where my interpretation of future events starts. To me an obvious follow up would incorporate small molecules as opposed to just other proteins. Getting proteins that bind well to specific ligands is still an open problem. Generally, this process starts in nature, by trying to find a naturally occurring protein with a desired functionality that might bind to a desired substrate. While tools like Bio-Prodict's 3DM were huge steps in making this process less arduous, it still requires human expertise.
Next, the protein needs to be optimized. Nature's best attempt for binding and/or converting a ligand of interest is likely not the best for industrial applications. Activity, specificity and stability are generally tuned for several rounds of guided evolution or (machine learning guided) rational design with the aim of getting several orders of magnitude increase in fit. These processes can take months and may not even yield a suitable design. If the high success rates reported for RFDiffusion hold up in this case, it would skip several steps in this process, lowering costs for designing custom proteins significantly.
Future Directions
I project that RFDiffusion will first be enhanced by replacing the Rosetta protein folding module with a more capable model. This could be AlphaFold2 or one of the high performers in CASP15 (Baker unfortunately did not rank in the top 10 this year). The paper notes explicitly that this is possible for an array of different models. Sergey Ovchinnikov already accomplished a hacked version of this on Twitter, less than a week after the initial pre-publication. Using AlphaFold2 as a design network may allow users to skip the AF2 validation step used by the authors for sequence verification. This is of course assuming that other models are not limited by lacking some of the design features RosettaFold exhibits.
Additional constraints outside a target protein could also be included. Guiding the diffusion process to produce structures more like a specific family of proteins would be a boon for enzyme design. Imagine specifying a target family of enzymes, a target ligand and letting the model rapidly design a protein that satisfies those constraints with high accuracy. Any number of pharmaceutical companies would be chomping at the bit to get access to this technology. RFDiffusion already is able to scaffold enzyme active sites without modeling small molecules explicitly. The authors demonstrate this capability in an experiment where they scaffold a retroaldolase active site.
Finally and most futuristically, this process could be used to design complex biological machinery. Incorporating the functionality of the protein and modeling that in some manner would allow for the design of an entire pipeline of proteins that construct specific complex molecules. Adding search based optimization for known conversions with different protein families would usher in an era of rapid biological process design. Just enter a resource and target molecule, let the servers run for a bit and test the resulting pathway to find your required complex molecule being made in the lab!

Conclusion
The Baker Lab has made significant strides in the space of protein design, with a great application of modern ML techniques combined with convincing experimental lab work. I trust that the very capable team is already working on developing this technique further, improving the accuracy and speed of this solution. In line with their previous projects, I expect that the source code for RFDiffusion will soon be published on GitHub under the BSD license. This will allow even more experimentation and innovative work to take place here.
Given the pace at which advances take place in the Machine Learning/Protein space, I have no doubt that we are on the cusp of a decade of rapid innovation. This will lead to giant leaps in pharmaceutical and industrial applications of proteins, resulting in improved health care and vastly more green and efficient manufacturing processes.
